
DiGeorge Syndrome: The Late Diagnosis That Makes Sense – Clinical Case Report
DOI:
https://doi.org/10.60591/crspmi.182Keywords:
DiGeorge Syndrome/diagnosis, DiGeorge Syndrome/genetics, DiGeorge Syndrome/therapyAbstract
DiGeorge syndrome is the most common microdeletion
syndrome and one of the velocardiofa-cial syndromes.
Congenital heart disease, primary hypoparathyroidism and
primary immunode-ficiency represent the classic triad of
manifestations. It is a very heterogeneous genetic pathol-
-ogy, with great phenotypic variability, with clinical features
and pleiotropic manifestations that can affect all organs and systems. Late diagnosis is still rare in adult medicine, with a high prob-ability of underdiagnosis. We present the case of a 33-year-old man with moderate cognitive delay and a history of epilepsy, primary hypoparathyroidism and schizophrenia admitted due to Evans syndrome. classic, namely primary hypoparathyroidism and primary immunodeficiency.
Hematological changes usually occur in childhood and are particularly rare in adulthood in patients with 22q11.2 microdeletion syndrome.
Downloads
References
Lambert MP, Arulselvan A, Schott A, Markham SJ, Crowley TB, Zackai EH, et al. The 22q11.2 deletion syndrome: Cancer predisposition, platelet abnormalities and cytopenias. Am J Med Genet A. 2018;176:2121-7. doi: 10.1002/ajmg.a.38474.
Al-taie N, Scheuter-Mlaker S, Schlesinger M, Abrahamian H. Case report: DiGeorge syndrome presenting with hypoparathyrodism and learning difficulties in adulthood. BJMP. 2014;7:a730.
Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, et al. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201-6. doi: 10.1016/0888-7543(91)90501-5.
Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813-7. doi: 10.1136/jmg.30.10.813.
McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23-9. doi: 10.1097/00125817-200101000-00006.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, el al. 22q11.2 deletion syndrome. Nat Rev Dis Primers.
;19:15071. doi: 10.1038/nrdp.2015.71.
Swillen A, McDonald-McGinn D. Developmental trajectories in 22q11.2 deletion. Am J Med Genet C Semin Med Genet. 2015;169:172-81. doi: 10.1002/ajmg.c.31435.
Fabiano RR, Zen PR, Graziadio C, Paskulin GA. Síndrome de deleção 22q11.2 e cardiopatias congênitas. Rev Paul Pediatr. 2011;29:251-60.
Lawrence S, McDonald-McGinn DM, Zackai E, Sullivan KE. Thrombocytopenia in patients with chromosome 22q11.2 deletion syndrome. J Pediatr. 2003;143:277-8. doi: 10.1067/S0022-3476(03)00248-8.
Kratz CP, Niehues T, Lyding S, Heusch A, Janssen G, Göbel U. Evans syndrome in a patient with chromosome 22q11.2 deletion syndrome: a case report. Pediatr Hematol Oncol. 2003;20:167-72. doi: 10.1080/0880010390158685.
Gupta S, Aggarwal S, Nguyen T. Increased spontaneous apoptosis in T lymphocytes in DiGeorge anomaly. Clin Exp Immunol. 1998;113:65-71. doi: 10.1046/j.1365-2249.1998.00629.x.
Oliveras-Cordero HA, Rivera-Jiménez E. Recurrent evans syndrome in a patient with 22q11.2 deletion syndrome: an uncommon hematological presentation. Cureus. 2020;12:e11510. doi: 10.7759/cureus.11510.
Mannering N, Hansen DL, Frederiksen H. Evans syndrome in children below 13 years of age - A nationwide population-based cohort study. PLoS One. 2020;15:e0231284. doi: 10.1371/journal.pone.0231284. 14. Brenner MK, Clarke S, Mahnke DK, Simpson P, Bercovitz RS, Tomita-Mitchell A, et al. Effect of 22q11.2 deletion on bleeding and transfusion utilization in children with congenital heart disease undergoing cardiac surgery. Pediatr Res. 2016;79:318-24. doi: 10.1038/pr.2015.216.
Bonati MT, Vanelli C, Sangalli D, Sina C, Giardino D, Sassone J, et al. Cerebral microbleeds: A new presenting feature of chromosome 22q11.2 deletion syndrome. J Neurol Sci. 2016;368:300-3. doi: 10.1016/j.jns.2016.07.044.
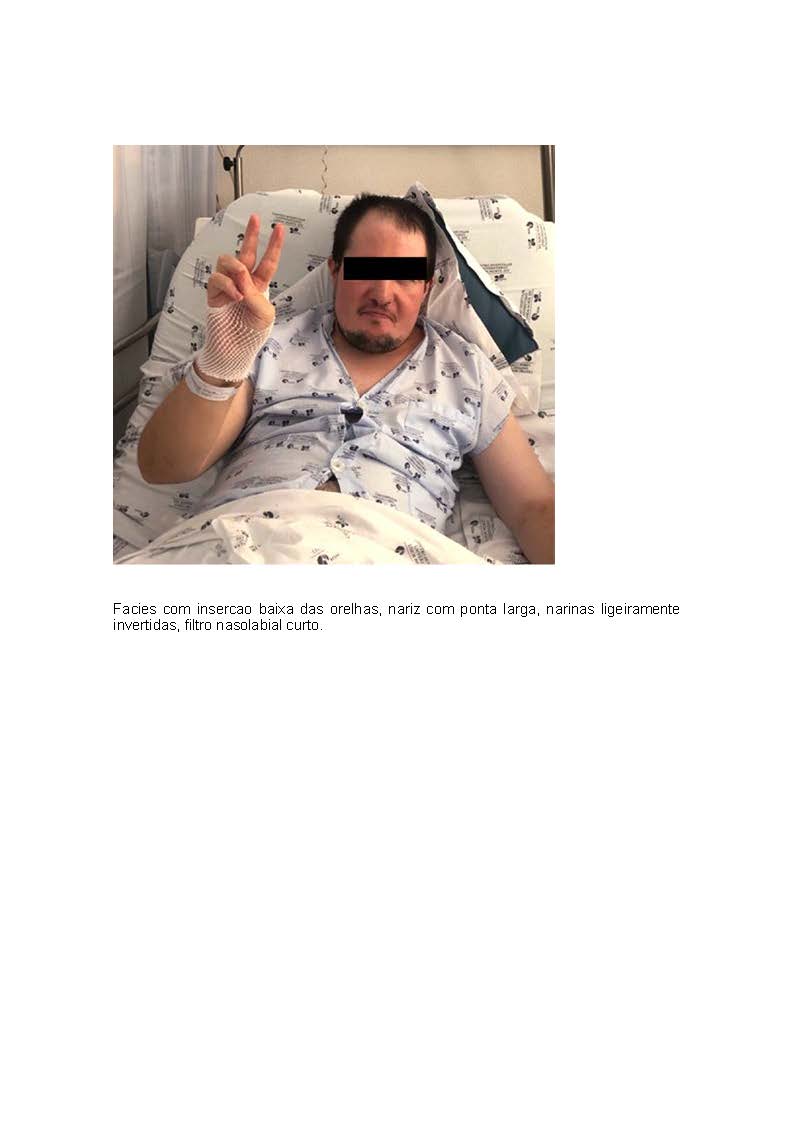
Downloads
Published
How to Cite
Issue
Section
Categories
License
Copyright (c) 2024 Bianca Cristea, Tiago Ferreira, Jorge Ferreira, Sandra D. Rebelo, Ana Tornada, Paula Alcântara

This work is licensed under a Creative Commons Attribution 4.0 International License.




